|
Dinucleotides
- Identification of the ATD Peaks
 By
comparing the cross sections of the theoretical structures to those determined
from the ATDs, each peak in the ATD can be identified in terms of a particular
conformation. By
comparing the cross sections of the theoretical structures to those determined
from the ATDs, each peak in the ATD can be identified in terms of a particular
conformation.
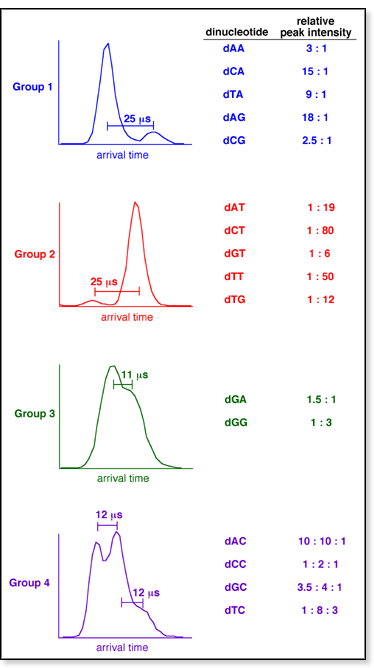
For Groups 1
and 2, the shortest-time peak can
be assigned as the stacked conformer and the longest-time peak
can be assigned as the open conformer. No significant amount of
the H-bonded conformers appears to be present for the dinucleotides
in these two groups.
In Group 3,
the shortest-time peak is consistent with the stacked conformer
and the longest-time peak is consistent with the H-bonded conformer.
In this case, the open conformer is not present.
In
Group 4, the shortest-time peak
can be assigned as the stacked conformer, the intermediate-time
peak as the H-bonded conformer, and the longest-time peak as the
open conformer.
Three
trends are apparent in the assignments of the ATD peaks:
- All three conformers are experimentally observed for only 4 of the
16 dinucleotides
- The stacked conformer is the only form observed for all 16
dinucleotides
- Conformational preferences are strongly dependent on the identity
of the 3' base
- Dinucleotides with a 3' A
favor stacked conformations
- Dinucleotides with a 3' C
favor H-bonded conformations
- Dinucleotides with a 3' T
favor open conformations
- Dinucleotides with a 3' G
depend on the identity of the 3' base
Base
stacking is usually thought to be favored only in solution. However, in
the case of the dinucleotides, it is the lowest-energy structure and readily
observed for all of the nucleotides. However, common Watson-Crick pairing
schemes that occur in double-stranded DNA is not possible in these dinucleotides.
Thus, stacking seems to be preferable.
The open conformer is experimentally observed
for all of the dinucleotides except dGA and dGG. Theoretical modeling
of these two dinucleotides indicates that the open conformers are
10 kcal/mol higher in energy than the stacked or H-bonded
conformers. For the other 14 dinucleotides, the open conformers
are predicted to be only 1-6 kcal/mol higher in energy.
The reason why the 3' base directs conformational
traffic is not clearly known. The calculations do not show any major structural
differences based on the 3' base. Ab initio calculations have indicated
that base stacking is stabilized by dispersion attractions (which are
not considered in our molecular mechanics/dynamics calculations) [Sponer,
J.; Leszczynski, J.; Hobza, P. Biopolymers 2001, 61,
3-31 and Hobza,
P.; Sponer, J. Chem. Rev. 1999, 99, 3247-3276].
Thus, higher-level theoretical modeling may be needed to understand the
role of the 3' base in determining conformational preferences.
|


