|
Zwitterions
- Bradykinin
 The
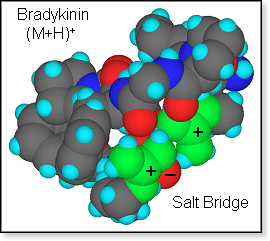
singly protonated nonapeptide bradykinin (BK) with the sequence arg-pro-pro-gly-phe-ser-pro-phe-arg
is ideally set up to form a salt bridge in the gas phase with both arginine
side chains protonated and the C-terminus deprotonated. However, from
cross section experiments in comparison with calculations
the zwitterion hypothesis can neither be confirmed nor rejected as both
salt bridge and charge solvation structures cover the same range of cross
sections.[17] The
singly protonated nonapeptide bradykinin (BK) with the sequence arg-pro-pro-gly-phe-ser-pro-phe-arg
is ideally set up to form a salt bridge in the gas phase with both arginine
side chains protonated and the C-terminus deprotonated. However, from
cross section experiments in comparison with calculations
the zwitterion hypothesis can neither be confirmed nor rejected as both
salt bridge and charge solvation structures cover the same range of cross
sections.[17]
The
large set of model structures obtained as part of the
cross section project was also used to attempt to answer the question:
Does gas-phase H/D-exchange data provide information about the gas-phase
structure of ions with labile hydrogen atoms? To address this question
we developed a simple model to predict the H/D-exchange kinetics for a
set of given model structures. Using our bradykinin molecular mechanics
structures obtained both for zwitterions and charge-solvation structures,
we found that a set of low-energy zwitterion structures matched the experimental
data obtained by others [18]
far better than a corresponding set of non-zwitterion structures.[14]
The
main features of the model we developed include:[14]
- D2O
samples entire peptide surface
- peptide
conformation does not change upon addition of D2O
- H/D-exchange
occurs by a "relay" mechanism,[19]
hence
- the
protonated site –AH+ has to be accessible to D2O
- a
"basic site" -B: has to be accessible to D2O
-
-B: must be close to –AH+.
- peptide
conformation does not change during collision time
- peptide
conformation may change on experimental time scale
To
check on some of the assumptions above we ran molecular dynamics calculations
on the (BK + H + D2O)+ system. We found that D2O
does move along the peptide surface, but that it does prefer to hang out
at certain locations on the peptide surface for extended periods of time.
Two of those locations, both near a protonated arginine side chain, are
indicated in the figure below for the example of a bradykinin salt bridge
structure.
|
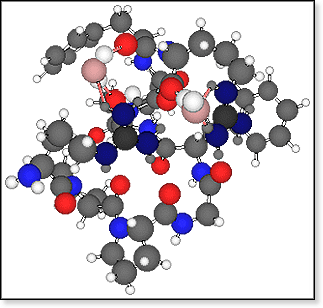 FIGURE
(LEFT): Two preferred sites of a water molecule binding to a doubly-protonated,
singly-deprotonated bradykinin molecule are shown. The water oxygen atom
is shown as a pink ball and the H-bonds to bradykinin as pink sticks. The
nitrogen atoms of the two charged guanidinium groups are shown in dark blue,
amide and amine nitrogens in light blue, bradykinin oxygen atoms in bright
red, carbon atoms in gray, and hydrogen atoms in white. FIGURE
(LEFT): Two preferred sites of a water molecule binding to a doubly-protonated,
singly-deprotonated bradykinin molecule are shown. The water oxygen atom
is shown as a pink ball and the H-bonds to bradykinin as pink sticks. The
nitrogen atoms of the two charged guanidinium groups are shown in dark blue,
amide and amine nitrogens in light blue, bradykinin oxygen atoms in bright
red, carbon atoms in gray, and hydrogen atoms in white. |
|
 Recent
hydration experiments carried out on protonated bradykinin
yield a pattern of water binding energies that is very different from
the protonated peptide LHRH, a decapeptide which cannot form a salt bridge
structure because it does not have any acidic site necessary for deprotonation
in the case of a zwitterion. Experimental water binding energies for the
first four water molecules adding to protonated bradykinin are nearly
identical, whereas for protonated LHRH the first water molecule is more
strongly bound than the second, which is in turn more strongly bound than
the third and fourth water molecule (see table above). Molecular mechanics
studies on these two systems including one to four water molecules indicate
quite different hydration properties for the two peptides in line with
the different patterns of hydration energies. For protonated LHRH the
water molecules solvate the peptide surface rather evenly with a slight
preference for adding the first water molecule to the protonated site. Recent
hydration experiments carried out on protonated bradykinin
yield a pattern of water binding energies that is very different from
the protonated peptide LHRH, a decapeptide which cannot form a salt bridge
structure because it does not have any acidic site necessary for deprotonation
in the case of a zwitterion. Experimental water binding energies for the
first four water molecules adding to protonated bradykinin are nearly
identical, whereas for protonated LHRH the first water molecule is more
strongly bound than the second, which is in turn more strongly bound than
the third and fourth water molecule (see table above). Molecular mechanics
studies on these two systems including one to four water molecules indicate
quite different hydration properties for the two peptides in line with
the different patterns of hydration energies. For protonated LHRH the
water molecules solvate the peptide surface rather evenly with a slight
preference for adding the first water molecule to the protonated site.
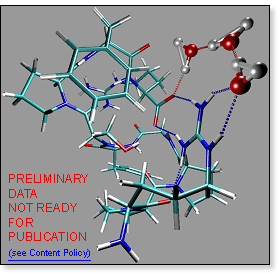 For
protonated bradykinin, on the other hand, water molecules prefer to form
a loop bound on both sides to the salt bridge: -COO-···H2O···H2O···H2O···H+Arg-. For
protonated bradykinin, on the other hand, water molecules prefer to form
a loop bound on both sides to the salt bridge: -COO-···H2O···H2O···H2O···H+Arg-.
It is reasonable to assume that inserting water molecules into the loop
lowers the system energy by a constant amount per water molecule inserted,
while the bradykinin conformation does not have to undergo any major changes.
This would explain perfectly well, why the first few water molecules have
nearly identical binding energies. Note, that flipping the bonds in the
loop converts the zwitterion into a charge solvation structure:
-COOH···OH2···OH2···OH2···Arg-
|


