|
Introduction
 Theoretical
computer calculations to determine the structures and energies of molecules
have been used for many years in the Bowers Group to help explain our
experimental observations. Initially these types of calculations were
restricted to relatively small systems
and consisted of ab initio and semiempirical molecular orbital calculations
on carbon clusters and transition metal complexes. In recent years, with
the advent of ionization techniques such as MALDI and ESI, our group has
begun to investigate larger chemical systems experimentally. To keep up
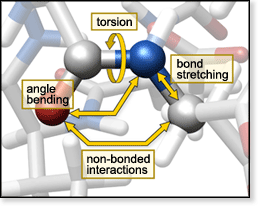
with these changes on the experimental side, we've adopted molecular mechanics
(MM) calculations as a theoretical method. MM calculates structures and
energies based on a force field determined by classical-mechanical motions
of the atoms in the molecule. The National Institutes of Health's Center
for Molecular Modeling has a helpful online
guide to MM which explains the principles behind the method. MM calculations
are much faster than ab initio and semiempirical molecular orbital calculations
and can be applied to much larger systems. This makes it possible to perform
dynamics calculations that track changes to molecular geometry over time
and temperature variations (see Structure Sampling).
Our group uses the AMBER
set of molecular mechanics/dynamics programs to investigate many systems
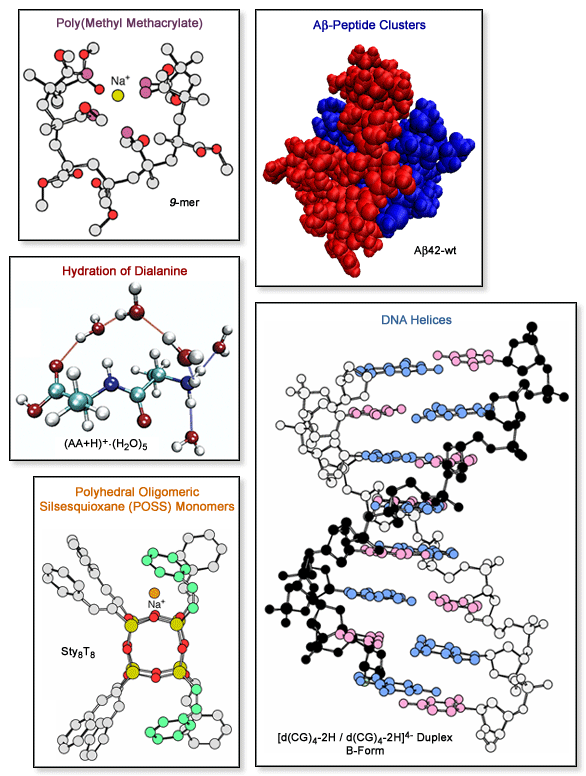
including synthetic polymers, biopolymers and materials. Several example
structures calculated by MM are shown below. We also collaborate with
Prof.
Joan-Emma Shea's theoretical/biophysical research group on modeling
projects. Theoretical
computer calculations to determine the structures and energies of molecules
have been used for many years in the Bowers Group to help explain our
experimental observations. Initially these types of calculations were
restricted to relatively small systems
and consisted of ab initio and semiempirical molecular orbital calculations
on carbon clusters and transition metal complexes. In recent years, with
the advent of ionization techniques such as MALDI and ESI, our group has
begun to investigate larger chemical systems experimentally. To keep up
with these changes on the experimental side, we've adopted molecular mechanics
(MM) calculations as a theoretical method. MM calculates structures and
energies based on a force field determined by classical-mechanical motions
of the atoms in the molecule. The National Institutes of Health's Center
for Molecular Modeling has a helpful online
guide to MM which explains the principles behind the method. MM calculations
are much faster than ab initio and semiempirical molecular orbital calculations
and can be applied to much larger systems. This makes it possible to perform
dynamics calculations that track changes to molecular geometry over time
and temperature variations (see Structure Sampling).
Our group uses the AMBER
set of molecular mechanics/dynamics programs to investigate many systems
including synthetic polymers, biopolymers and materials. Several example
structures calculated by MM are shown below. We also collaborate with
Prof.
Joan-Emma Shea's theoretical/biophysical research group on modeling
projects.
|