|
Conformational
Sampling
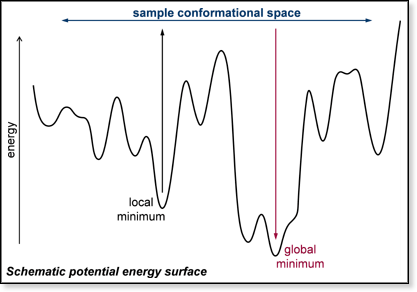 In
many of our molecular modeling investigations the goal is to determine
the lowest-energy conformation of a molecule. The larger the molecule
is, the more difficult this becomes. The schematic potential energy surface
at right illustrates the concept. Large molecules have a large number
of possible conformations, many of which represent local minima on the
potential energy surface. The goal of the calculation is to find low-energy
minima including the global minimum. The first step in a MM calculation
is to obtain an initial molecular structure. Starting structures are often
built on the computer using the AMBER
software or another modeling program called HyperChem.
For large molecules it is useful to start with a known structure obtained
by some other method, such as X-ray crystallography. These structures
can often be obtained from the Protein
Data Bank, a collection of 3-D biological molecular structure data.
If the procedure used to sample the molecule's conformational space is
adequate, the results of the MM calculation should not depend on the choice
of starting structure. After obtaining an initial structure, the next
step is to optimize its geometry by minimizing its energy. The resulting
conformation corresponds to a local minimum on the potential energy surface. In
many of our molecular modeling investigations the goal is to determine
the lowest-energy conformation of a molecule. The larger the molecule
is, the more difficult this becomes. The schematic potential energy surface
at right illustrates the concept. Large molecules have a large number
of possible conformations, many of which represent local minima on the
potential energy surface. The goal of the calculation is to find low-energy
minima including the global minimum. The first step in a MM calculation
is to obtain an initial molecular structure. Starting structures are often
built on the computer using the AMBER
software or another modeling program called HyperChem.
For large molecules it is useful to start with a known structure obtained
by some other method, such as X-ray crystallography. These structures
can often be obtained from the Protein
Data Bank, a collection of 3-D biological molecular structure data.
If the procedure used to sample the molecule's conformational space is
adequate, the results of the MM calculation should not depend on the choice
of starting structure. After obtaining an initial structure, the next
step is to optimize its geometry by minimizing its energy. The resulting
conformation corresponds to a local minimum on the potential energy surface.
Most often we are interested in the global minimum
and/or the lowest-energy local minima. To find those stable conformations,
we use a technique called simulated annealing.
In this procedure we increase the internal energy of the molecule and
allow the starting structure to change accordingly. By reminimizing the
energy of the resulting structure, a new stable conformation can be found.
By repeating this procedure the global minimum (lowest-energy) structure
can ultimately be found. The details of this sampling process can vary.
Sometimes it is also useful to examine the change
of shape as a consequence of thermal motion for comparison with experiment,
which is typically carried out at room temperature. In these instances
molecular dynamics calculations are done to sample conformation space
at a specific temperature.
For investigations of large molecules (such as
the Aβ peptides) conducted in collaboration
with the Shea
Group, we use the replica exchange technique in order to sample conformational
space efficiently at a given temperature. This technique takes both energetic
and entropic effects into account.
|


