|
Simulated
Annealing
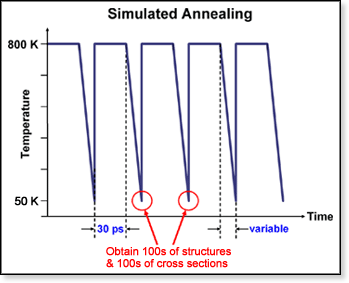 The
simulated annealing procedure is used to generate a group of candidate
molecular structures by molecular mechanics. The cross sections of these
theoretical structures can then be compared with experimental results.
(The method used for obtaining cross sections from theoretical molecular
structures is detailed in the section on Theoretical
Collision Cross Sections.) The structures are generated using the
AMBER suite of
programs by subjecting a starting conformation to 30 ps of dynamics at
800 K, then cooling it to 50 K over a variable period of time. From there,
the molecule is geometry-optimized by minimizing its energy. This first
candidate structure is then used as a new input conformation and subjected
to the same annealing sequence. In this fashion typically 100 candidate
structures are generated in an attempt to cover reasonable conformations
for a given system. This procedure is illustrated in the figure above.
The results of the annealing process are often presented as a scatter
plot of cross section versus relative energy. An
example of a scatter plot is shown below for the dinucleotide (dGC-H)-.
Each point represents a theoretical structure obtained in the The
simulated annealing procedure is used to generate a group of candidate
molecular structures by molecular mechanics. The cross sections of these
theoretical structures can then be compared with experimental results.
(The method used for obtaining cross sections from theoretical molecular
structures is detailed in the section on Theoretical
Collision Cross Sections.) The structures are generated using the
AMBER suite of
programs by subjecting a starting conformation to 30 ps of dynamics at
800 K, then cooling it to 50 K over a variable period of time. From there,
the molecule is geometry-optimized by minimizing its energy. This first
candidate structure is then used as a new input conformation and subjected
to the same annealing sequence. In this fashion typically 100 candidate
structures are generated in an attempt to cover reasonable conformations
for a given system. This procedure is illustrated in the figure above.
The results of the annealing process are often presented as a scatter
plot of cross section versus relative energy. An
example of a scatter plot is shown below for the dinucleotide (dGC-H)-.
Each point represents a theoretical structure obtained in the  simulated annealing process. The structures obtained in this process can
often be grouped into families with similar structural features. Conformations
within 5 kcal/mol of the lowest-energy conformation are generally considered
reasonable models.
simulated annealing process. The structures obtained in this process can
often be grouped into families with similar structural features. Conformations
within 5 kcal/mol of the lowest-energy conformation are generally considered
reasonable models.
The cooling step in the annealing protocol is
an important one. The temperature drop has to occur sufficiently slowly
to ensure appropriate equilibrium of the system. Since the equilibration
time is temperature-dependant with rapid equilibration at high temperatures
and slow equilibration at low temperatures, an exponential temperature
profile is most suitable for locating the global minimum. However, in
the past we have repeatedly found experimental evidence for the existence
of structures other than the AMBER global minimum. Hence, locating reasonable
theoretical structures above the global minimum is often useful for the
interpretation of experimental data. Therefore, full equilibration during
the cooling step is not always desired, and we found that a linear temperature
profile from 800 K to 50 K does a reasonable job for our purposes (see
"linear profile sampling procedure" below).
The simulated annealing approach with a linear
temperature profile during the cooling step has been evaluated using the
oligoglycine system of small peptides. The method appears to cover conformational
space fairly well for molecules of this size. Details of the evaluation
are given below, but as an example, protonated pentaglycine was found
to have 28 different conformers (out of 100 attempts) spread over an energy
range of about 15 kcal/mol, 9 of them within the lowest 5 kcal/mol. Thus
each conformation was found more than 3 times on average. However, the
low-energy conformations were located much more frequently, for instance,
the one with the lowest energy 25 times.
Evaluation
of the "linear profile sampling procedure". The simulated
annealing procedure has been used in our group on a number of systems
including crown ethers, oligosaccharides, synthetic polymers and peptides
such as bradykinin. To gauge how well this method leads to the desired
molecular structures, we have performed an in-depth analysis of the small
peptide systems GlynH+ and GlynNa+
for n = 1-6. The concept of simulated annealing has clearly not
been developed with the intention of covering the entire conformational
space of a molecule, but rather as a means of searching for the global
minimum on a potential energy surface. The method obviously discriminates
against higher-energy conformers. Therefore, we do not expect to find
all possible minima on the potential energy surface, but rather those
deep wells that are sufficiently distant from the nearest deeper
well and separated from it by a substantial barrier. It is the conformers
associated with these minima in which we are most interested.
For each of the systems GlynH+
(with protonated N-terminus), Glyn(cs)Na+
(with Glyn in a non-zwitterion charge solvation structure),
and Glyn(zw)Na+ (with a Glyn
zwitterion), one hundred low-energy conformers were determined using AMBER
as described above. For each species investigated, some of the 100 conformers
are identical. We are therefore interested in how many distinct conformations
are found as well as whether the lowest-energy conformation is the one
most frequently located. A table summarizing this type of data for the
oligoglycine systems is shown below. The data show, not surprisingly,
that the number of distinct conformers found increases with 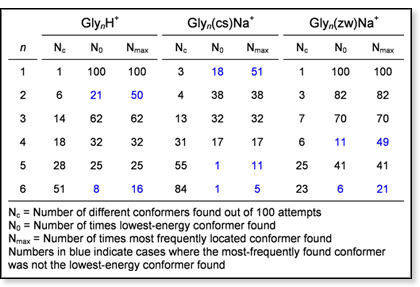 increasing
system size and that it approximately doubles for each additional glycine
unit. In almost all cases, the lowest-energy conformation located was
found more than five out of 100 attempts. The only exceptions are Gly5(cs)Na+
and Gly6(cs)Na+. In these cases, many slightly
different conformers with very similar energies were found. In general,
the lowest-energy conformer is also the one found most frequently. However,
there are a few exceptions, indicating that not only the depth of a well,
but also the size of the area in conformational space leading into the
well is a determining factor for the probability of its detection. A striking
example of this is the Gly5(cs)Na+ system: while
the lowest-energy conformation was found only once out of 100 structures,
another
more extended conformation with an energy ~1.5 kcal/mol above that of
the lowest-energy conformation is located more than 10 times. increasing
system size and that it approximately doubles for each additional glycine
unit. In almost all cases, the lowest-energy conformation located was
found more than five out of 100 attempts. The only exceptions are Gly5(cs)Na+
and Gly6(cs)Na+. In these cases, many slightly
different conformers with very similar energies were found. In general,
the lowest-energy conformer is also the one found most frequently. However,
there are a few exceptions, indicating that not only the depth of a well,
but also the size of the area in conformational space leading into the
well is a determining factor for the probability of its detection. A striking
example of this is the Gly5(cs)Na+ system: while
the lowest-energy conformation was found only once out of 100 structures,
another
more extended conformation with an energy ~1.5 kcal/mol above that of
the lowest-energy conformation is located more than 10 times.
It is important to consider whether the trends
observed for these relatively small oligoglycine compounds will carry
over to larger systems. The data in the table above suggests that a thorough
search for the global minimum should include at least 100 annealing cycles
for hexaglycine and should be doubled for each additional glycine unit.
However, in many cases, the exact location of the global minimum is not
required, but tracking down a local minimum near the global minimum is
sufficient. We have to keep in mind that the potential energy surface
itself is described by a rather crude model. Thus, the global minimum
of the model is not necessarily expected to represent the true lowest-energy
conformation anyway. Furthermore, one single zero-Kelvin structure is
not a very useful comparison with any experimental data, usually obtained
at T >> 0 K. Therefore a coarser search, cutting down computational
effort by a factor of 2 to 10 from the guidelines given above, can be
justified. If desired, the model global minimum could be found rapidly
by an additional local search around the deepest local wells.
|


