
 |

|
|||||
|
Theoretical Dynamics Data for PEG, PPG, and PTMEG - obtained using molecular mechanics methods |
|||||
For Na+PPG5, the maximum variance in cross-section is ±5 Å2 throughout the entire dynamics run. There is also a slight, steady decrease in cross-section from 300 K to 600 K that follows the trend observed in the experimental data. For Na+PTMEG5, the maximum variance in cross-section is ±10 Å2 at 600 K (twice that of Na+PPG5). The average cross-section remains essentially constant from 400 K to 600 K, again in agreement with the trend observed in the experimental data. In both of these oligomers, all 6 available oxygens remain coordinated to the Na+ ion throughout the entire dynamics run, essentially holding the oligomer in place. The only difference is in the increased motion involved with the "ring" surrounding the Na+ ion. For Na+PPG14 and Na+PTMEG11, the maximum spread in cross-section increases from ~±10 Å2 at 200 K to ±27 Å2 (for Na+PPG14) and ±35 Å2 (for Na+PTMEG11) at 600 K. The average cross-section also increases steadily from 400 K to 600 K. In these simulations, the part of the oligomer that is coordinated to the Na+ ion remains in place but the part that is remote from the Na+ ion unravels at higher temperatures, thus increasing the average "size" or cross-section of the oligomer. 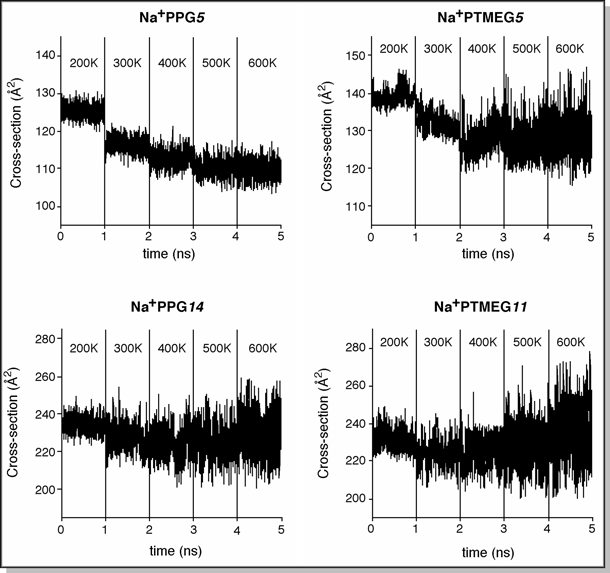 |
|||||