|
 The
CID spectra of M+PSn exclusively shows small M+-fragment
ions containing each end group of the oligomer (A1, A1,
A1, B1, B1, B1, see CID
Results Section) but the theoretical results show no evidence that
the M+ cation prefers to be near the ends of the oligomer.
So, the question is how are small A1, A1 and B1,
B1 fragments formed? The
CID spectra of M+PSn exclusively shows small M+-fragment
ions containing each end group of the oligomer (A1, A1,
A1, B1, B1, B1, see CID
Results Section) but the theoretical results show no evidence that
the M+ cation prefers to be near the ends of the oligomer.
So, the question is how are small A1, A1 and B1,
B1 fragments formed?
The
most likely answer, and one that has been published (see the references
listed in the CID Results Section), is
that the PS oligomer is randomly cleaved along the backbone and then rapidly
depolymerizes to each end. This mechanism was also proposed for PMMA
but the theoretical results for this system indicated that the M+
cation preferred to remain with larger fragments and so depolymerization
would be an energetically uphill battle. The M+PMMAn
structures were also determined to be U-shaped which led to another possible
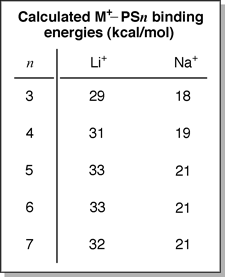
fragmentation mechanism. For M+PSn, there is no energetic
preference for M+ to remain attached to larger fragments as
seen in the calculated M+-PSn binding energies. This
consistency in binding energy is most likely due to the fact that the
metal cation only interacts with two phenyl groups and any additional
styrene units has little effect on the M+-PSn interaction.
Thus, each depolymerization step is likely to be thermoneutral and high-energy
collisions with the buffer gas during the CID experiments should be more
than enough to induce successive depolymerization steps.
|


