|
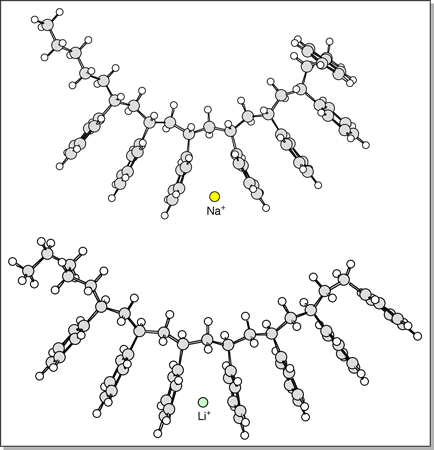 The
lowest-energy structure found for the PS 7-mer cationized by Li+
and Na+ are shown at right. The oligomers are quasi-linear
with the phenyl groups aligned on the same side of the PS backbone and
the M+ ion sandwiched between two phenyl groups near the middle
of the chain. Similar structures were predicted for the 3-mer to
6-mer. The only difference between the Li+PSn
and Na+PSn structures is that the larger Na+
ion pushes the two adjacent phenyl groups farther apart than Li+.
The average distance between M+ and the central point within
a neighboring phenyl group is 2.0 Å for Li+ and 2.4 Å
for Na+. (This may account for the slight increase in cross-section
observed for the PS 3-mer and 4-mer when Na+ is used
as the cation vs. Li+. For higher n, the overall size
of the oligomer apparently outweighs this small structural difference,
as the cross-sections for the 5-mer to 7-mer are similar
for Na+ and Li+.) The calculated cross-sections
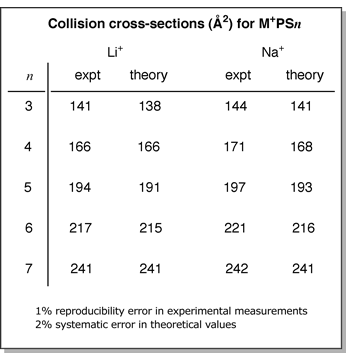
of these quasi-linear structures agree very well (within 2%) with experimental
values as shown in the table below. The
lowest-energy structure found for the PS 7-mer cationized by Li+
and Na+ are shown at right. The oligomers are quasi-linear
with the phenyl groups aligned on the same side of the PS backbone and
the M+ ion sandwiched between two phenyl groups near the middle
of the chain. Similar structures were predicted for the 3-mer to
6-mer. The only difference between the Li+PSn
and Na+PSn structures is that the larger Na+
ion pushes the two adjacent phenyl groups farther apart than Li+.
The average distance between M+ and the central point within
a neighboring phenyl group is 2.0 Å for Li+ and 2.4 Å
for Na+. (This may account for the slight increase in cross-section
observed for the PS 3-mer and 4-mer when Na+ is used
as the cation vs. Li+. For higher n, the overall size
of the oligomer apparently outweighs this small structural difference,
as the cross-sections for the 5-mer to 7-mer are similar
for Na+ and Li+.) The calculated cross-sections
of these quasi-linear structures agree very well (within 2%) with experimental
values as shown in the table below.
 The
lowest energy structures predicted for Li+PSn and Na+PSn
indicate that the metal cation prefers to be near the middle of the chain
but if the metal cation is positioned randomly along the chain, the overall
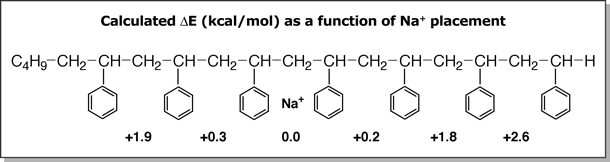
energy of the oligomer does not drastically change, as shown below. The
lowest energy structures predicted for Li+PSn and Na+PSn
indicate that the metal cation prefers to be near the middle of the chain
but if the metal cation is positioned randomly along the chain, the overall
energy of the oligomer does not drastically change, as shown below.
For example, when Na+ is sandwiched
between two phenyl groups near the ends of the PS oligomer, the resulting
structure is predicted to be 1.9 kcal/mol and 2.6 kcal/mol higher in energy.
However, the overall cross-section of the oligomer increases when the
metal cation is moved towards the ends of the oligomer because the phenyl
groups that are farther away from the metal cation do not line up as well
as when the metal cation is near the middle of the chain.
In commercial poly(styrene) the phenyl groups
are usually randomly oriented about the PS backbone (atactic) rather than
lined up on one side of the backbone as in the Li+PS and Na+PS
structures (isotactic). If the metal cation is removed, theory predicts
a much more random structure as seen below (bottom) for the lowest energy
structure of neutral PS7.
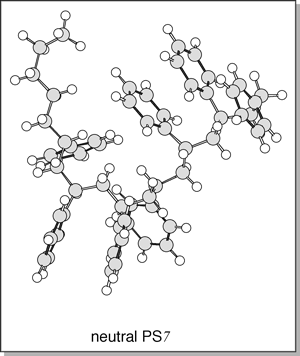 Although
neighboring phenyl groups form stacked pairs in the neutral species, each
pair is not aligned with any other stacked pair. The calculated cross-sections
of the neutral PSn structures are ~8% smaller than the Li+PSn
or Na+PSn structures. Thus, atactic structures are not
likely present in the cationized systems (at least in the smaller oligomers
shown here). This could have significant consequences as the tacticity
of polymers greatly influence their thermal and chemical properties. Although
neighboring phenyl groups form stacked pairs in the neutral species, each
pair is not aligned with any other stacked pair. The calculated cross-sections
of the neutral PSn structures are ~8% smaller than the Li+PSn
or Na+PSn structures. Thus, atactic structures are not
likely present in the cationized systems (at least in the smaller oligomers
shown here). This could have significant consequences as the tacticity
of polymers greatly influence their thermal and chemical properties.
|


