|
 Molecular
mechanics/dynamics calculations were used to generate trial structures
of the M+PMMA oligomers. The lowest-energy structures found
for the PMMA 5-mer, 7-mer, and 9-mer cationized by
Na+ are shown at right. The oligomers are U-shaped with the
sodium cation exclusively bound to carbonyl oxygens near both ends of
the oligomer (shown in purple). Similar low-energy structures were also
found for PMMA oligomers cationized by Li+, K+,
Rb+ and Cs+. Molecular
mechanics/dynamics calculations were used to generate trial structures
of the M+PMMA oligomers. The lowest-energy structures found
for the PMMA 5-mer, 7-mer, and 9-mer cationized by
Na+ are shown at right. The oligomers are U-shaped with the
sodium cation exclusively bound to carbonyl oxygens near both ends of
the oligomer (shown in purple). Similar low-energy structures were also
found for PMMA oligomers cationized by Li+, K+,
Rb+ and Cs+.
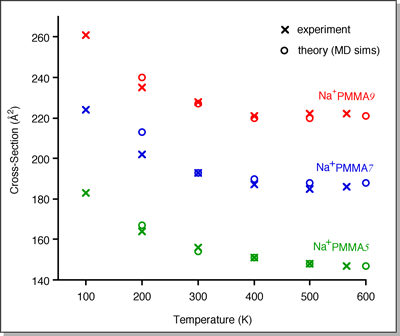
The average cross-sections of the U-shaped structures
are compared to the 300 K experimental values in the plot below (for Na+PMMAn).
Theory and experiment agree within 1-2%.
In the temperature-dependent experiments, the cross-sections
for Na+PMMAn did not increase at higher temperatures
indicating the oligomers are not unraveling. This makes sense based on
the U-shaped structures predicted by theory. The Na+ ion, which
binds to oxygens near each end of the oligomer, effectively acts as an
anchor and limits the amount of thermal motion the PMMA oligomer can undergo.
Molecular dynamics simulations on Na+PMMAn are consistent
with this interpretation. The dynamics were begun at 200 K and run for
1000 ps. Every 1 ps the structure was saved and its cross-section calculated.
At the end of the dynamics run, the temperature is increased 100 K and
the process repeated. Shown below (second from bottom) is the resulting
cross-section vs. time and temperature plot for the MD simulations of
Na+PMMA9. The maximum spread in cross-section at 600
K is only ±15 Å2.  For
a similar sized Na+PEG oligomer, which unraveled at 600 K,
the maximum spread in cross-section at 600 K was ±30 Å2. For
a similar sized Na+PEG oligomer, which unraveled at 600 K,
the maximum spread in cross-section at 600 K was ±30 Å2.
The average cross-section at each temperature in the
MD simulations is compared to the experimental values in the plot below
(bottom). The trend in cross-section shown in the experiments is nicely
reproduced by theory.
|