
|
|
|
 |
|
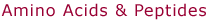 |
|
|
Oligoglycine
Appendix
|
|
|
Gly1
 Structure
(a) in the figure at right corresponds to the most stable conformer of
GlyH+ obtained by DFT calculations using a B3LYP/6-311G** basis
set. The dihedral angle α is 0° [see depiction
in (b)], but rotation about the C-N axis does not appear to be severely
hindered, since the conformation with α=60°
is only 1 kcal/mol higher in energy. For GlyNa+ two structures
are possible, one with the sodium ion bound to neutral glycine in its
amino acid form [structure (c)], and one with Na+ bound to
zwitterionic glycine [structure (d)]. A full geometry optimization on
a MP2/6-311++G** level indicates, that the first form (c) is some 3.6
kcal/mol more stable than the second one (d) after zero point energy correction.
The much faster DFT B3LYP/6-311++G**calculation agrees with the MP2 result
within 0.3 kcal/mol. The structures found by molecular
modeling are with minor deviations the same as the ab initio conformations.
The single minima located for GlyH+ (α=60°)
and GlyNa+ (zwitterion; α=0°) correspond
to the structures shown above. For Na+ bound to Gly in its
amino acid form three minima were found, the lowest one being that shown
in structure (c) above. Structure
(a) in the figure at right corresponds to the most stable conformer of
GlyH+ obtained by DFT calculations using a B3LYP/6-311G** basis
set. The dihedral angle α is 0° [see depiction
in (b)], but rotation about the C-N axis does not appear to be severely
hindered, since the conformation with α=60°
is only 1 kcal/mol higher in energy. For GlyNa+ two structures
are possible, one with the sodium ion bound to neutral glycine in its
amino acid form [structure (c)], and one with Na+ bound to
zwitterionic glycine [structure (d)]. A full geometry optimization on
a MP2/6-311++G** level indicates, that the first form (c) is some 3.6
kcal/mol more stable than the second one (d) after zero point energy correction.
The much faster DFT B3LYP/6-311++G**calculation agrees with the MP2 result
within 0.3 kcal/mol. The structures found by molecular
modeling are with minor deviations the same as the ab initio conformations.
The single minima located for GlyH+ (α=60°)
and GlyNa+ (zwitterion; α=0°) correspond
to the structures shown above. For Na+ bound to Gly in its
amino acid form three minima were found, the lowest one being that shown
in structure (c) above.
|
|
|
Gly2
 Molecular
mechanics calculations indicate that diglycine ionized at the N-terminus,
Gly2H+ [structure (a) in figure at right] and Gly2Na+
[structure (b)], prefer a cyclic arrangement as depicted for structure
(a). The corresponding extended linear conformations are ~7 kcal/mol higher
in energy. Note, that the dihedral angle ω
along the peptide bond, which usually is ~180° ("trans") in any oligoglycine
or other peptide systems, is ~0° ("cis") for the lowest-energy conformers
in both of these dimer systems. In this arrangement the protonated N-terminus
can be better solvated by the C-terminal oxygen. The sodiated glycine
form (c) is determined by the solvation of Na+ by the two >C=O
groups. The terminal -NH2 is either also bound to Na+
or forms a hydrogen bond with the amide hydrogen as shown in (c), the
latter structure being ~1.5 kcal/mol more stable. DFT calculations on
Gly2Na+ indicate at all levels of theory, that form
(c) is more stable by more than 10 kcal/mol than form (b). Molecular
mechanics calculations indicate that diglycine ionized at the N-terminus,
Gly2H+ [structure (a) in figure at right] and Gly2Na+
[structure (b)], prefer a cyclic arrangement as depicted for structure
(a). The corresponding extended linear conformations are ~7 kcal/mol higher
in energy. Note, that the dihedral angle ω
along the peptide bond, which usually is ~180° ("trans") in any oligoglycine
or other peptide systems, is ~0° ("cis") for the lowest-energy conformers
in both of these dimer systems. In this arrangement the protonated N-terminus
can be better solvated by the C-terminal oxygen. The sodiated glycine
form (c) is determined by the solvation of Na+ by the two >C=O
groups. The terminal -NH2 is either also bound to Na+
or forms a hydrogen bond with the amide hydrogen as shown in (c), the
latter structure being ~1.5 kcal/mol more stable. DFT calculations on
Gly2Na+ indicate at all levels of theory, that form
(c) is more stable by more than 10 kcal/mol than form (b).
|
|
|
GlynH+,
n=3-6
Semiempirical
geometry optimizations carried out on structures obtained from molecular
mechanics calculations for Gly4H+ and Gly5H+
indicate that salt bridge structures of the form I are substantially less
stable than simple charge solvation structures of type II.
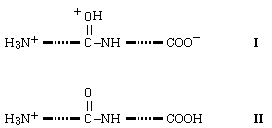
Thus,
for energetic reasons protonated oligoglycines of the form I can be excluded
as possible candidate structures not only for the small Gly2H+
and Gly3H+, but also when the chain is long enough
to allow separation of the two positive charges and formation of a salt
bridge between the positive N-terminus and the negative C-terminus. Molecular
mechanics results indicate that protonated oligoglycines prefer a
cyclic arrangement where one of the N-terminal amino hydrogens forms a
hydrogen bond to the C-terminal >C=O group (see figure below). Some
of the amide >C=O groups are also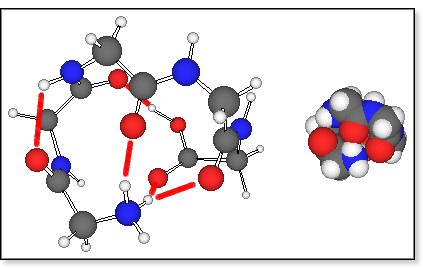 bound to the N-terminus to help solvate the charge. The lowest-energy
conformers are held together by three such H-bonds, which fold the peptide
into a pretty spherical shape. More extended conformers are typically
higher in energy and there is a reasonable correlation between the energy
of a conformation and its calculated cross section or degree of unfolding.
bound to the N-terminus to help solvate the charge. The lowest-energy
conformers are held together by three such H-bonds, which fold the peptide
into a pretty spherical shape. More extended conformers are typically
higher in energy and there is a reasonable correlation between the energy
of a conformation and its calculated cross section or degree of unfolding.
|
|
|
GlynNa+,
n=3-6
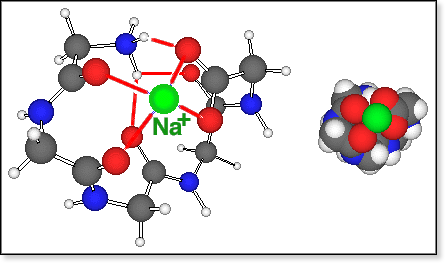
The
GlynNa+ structures (where Glyn is a non-zwitterion)
obtained by molecular
mechanics are determined by the solvation of Na+ by >C=O.
Even for the hexamer, all >C=O groups can coordinate around the Na+
ion, but the lowest-energy conformers (for n=4-6) have 4 to 5 such electrostatic
O-Na bonds. This is still a large coordination number and combined with
the rather long O-Na bonds of 2.3 to 2.5 Å, it causes the relatively
small peptide to unfold and span around Na+ like an open umbrella,
resulting in a quite oblate shape (see figure below).
The
conformers of zwitterionic oligoglycine bound to Na+ are all
fairly compact. In all cases we were able to locate only a small number
of different conformers using our simulated annealing procedure, and in
all cases there exists one particularly stable conformation, shown in
the figure below for Gly5Na+. All of those "salt
bridge" conformers have in common that both Na+ and -NH3+
are bound to the carboxylate group via an electrostatic bond and 1 or
2 H-bonds, respectively. The amide >C=O groups coordinate either to
Na+ or to -NH3+. The coordination number
for Na+ is typically 4 (3 for n=3), and
-NH3+ forms between 2 (for n=3,4) to 3 (for n=6)
hydrogen bonds.
|
|
