Background. Intermolecular interactions are at the heart of virtually all chemical or biochemical phenomena. The computational area often known as "molecular docking" explores the interaction of molecules, usually by taking into account in some way the energetics of these interactions, as well as the conformations of interacting partners. For any docking study, it is necessary to define somehow a receptor and a molecule to interact with that receptor. The tertiary structure of the receptor can be determined by crystallographic or NMR methods or can be inferred from the structures of related molecules by "homology modeling". A complication in computer-based docking is that the conformations of both the receptor and the ligand in their complex may be different from their conformation(s) when they are free of each other ("induced fit").
Molecular docking is an important aspect of efforts to find "lead compounds" for the development of new drugs. In a typical application of this technology, an array of possible ligands for the receptor is screened computationally in order to select a few compounds for more detailed scrutiny. The group of ligand screened can simply be all of the compounds in a database or catalog of reagents. If some details of a binding interaction are known, it may be possible to pre-select ligands so that they have a particular shape or characteristic distribution of electrical charges.
There are tutorials for manually docking one molecule into another available in SYBYL (Look under DOCK.) For those with good hand-eye coordination (and plenty of video game experience) manual docking is a productive way to explore the interaction of a specific ligand with a receptor. However, this procedure is too time-consuming if thousands of compounds have to be screened.
A number of computer programs for docking one molecule into another have been developed and made available at little or no charge to academic institutions. Among these are DOCK (http://www.cmpharm.ucsf.edu/kuntz/dock.html), AutoDock (http://www.scripps.edu/pub/olson-web/doc/autodock), FlexX (http://cartan.gmd.de/flexx/) and FlexiDock (http://www.tripos.com/ software/fdock.html). DOCK and AutoDock have been licensed and installed on the CCBL system. FlexX can be downloaded and installed in a form that will be usable for about six months. (This has not been done for the CCBL system but may be done by individual users.) FlexiDock is licensed and documented as part of the SYBYL package; look under the Biopolymer menu. (There is a Tripos tutorial available for Flexidock.) Copies of printed documentation for all of these programs are available on the shelf at the front of Rm 1153.
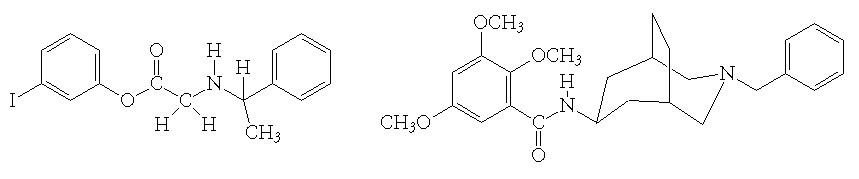
This exercise. The sulfhydryl protease papain has an active site that features an elongated groove with a pocket at each end, with the pockets separated by a ridge. A panel of molecules that have shapes that are or could be complementary to this binding site and, thus, could be inhibitors of the enzyme, was selected by screening the entire Cambridge Crystallographic Database. Two of the molecules that could be ligands for the enzyme are shown below. Determine computationally whether or not these molecules can be inhibitors of papain and predict which one of them will have the smallest binding constant.
Suggestions and comments.
[1] Familiarize yourself with the shape of the active site of papain and of the two prospective ligands. Form an opinion of how these molecules are likely to fit into the active site of the protein.
[2] Please note that the two prospective inhibitors are flexible in their middle sections. This flexibility could help the ligand adopt to the shape of the protein binding site.
[3] In answering the question posed, you will possibly end up using one of the programs mentioned above. While these programs have been installed and some documentation printed, no additional information about them will be made available. You will have to become familiar with the operation of the program chosen and battle with making it do what you want it to do. There is some assistance available at the respective WWW sites and the authors of the programs have published extensively on the applications of their programs. Be aware that most of the programs have no GUI and will be run in stand-alone/batch mode.
[4] Some investigation of the background of the enzyme, its inhibition and computational docking methods before starting the project may save some time ("An hour in the library is worth a day in the lab.")
[5] Please return any manuals borrowed to the lab when you are finished with them.
[6] The report for this exercise is due the last day of class, March 15. The report should include a short discussion of the chemistry, structure and inhibition of the enzyme (references to review articles are fine), a discussion of your strategy for solving the problem, your results, and a discussion of the probable reliability of your results.