|
Prion Proteins/TSEs - Introduction
 Transmissible spongiform encephalopathies (TSEs) are a family of diseases that infect many mammalian species including humans (Creutzfeldt-Jakob disease), sheep and goats (scrapie), cattle (bovine spongiform encephalopathy, BSE, commonly referred to as “mad cow” disease), and deer and elk (chronic wasting disease). TSEs have become increasingly prevalent in recent years. An outbreak of hundreds of thousands of cases of the bovine form, BSE, occurred in Britain in the 1980s and here in the United States, chronic wasting disease is becoming a serious threat to our deer and elk populations. BSE is unusual in that it has crossed the species barrier and infected and killed more than 100 people in the UK, including two people who recently contracted the disease from blood transfusions. Transmissible spongiform encephalopathies (TSEs) are a family of diseases that infect many mammalian species including humans (Creutzfeldt-Jakob disease), sheep and goats (scrapie), cattle (bovine spongiform encephalopathy, BSE, commonly referred to as “mad cow” disease), and deer and elk (chronic wasting disease). TSEs have become increasingly prevalent in recent years. An outbreak of hundreds of thousands of cases of the bovine form, BSE, occurred in Britain in the 1980s and here in the United States, chronic wasting disease is becoming a serious threat to our deer and elk populations. BSE is unusual in that it has crossed the species barrier and infected and killed more than 100 people in the UK, including two people who recently contracted the disease from blood transfusions.
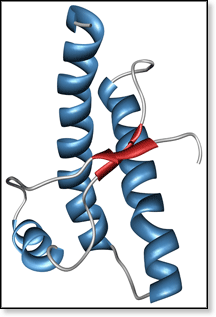
BSE belongs to a family of neurological diseases caused by misfolding and aggregation of proteins. It occurs when a common mammalian protein PrPC (prion protein cellular) misfolds to form PrPSc (prion protein scrapie). The solution-phase NMR structure [1] of a PrPC segment is shown in the figure above. Evidence suggests that native PrPC is recruited by misfolded PrPSc to form a dimer complex, which rearranges to form (PrPSc)2. This scrapie dimer can then either dissociate to regenerate PrPSc or aggregate to form fibrils. A proposed mechanism is shown below.
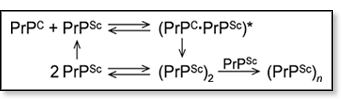 There is growing evidence that the disease is both transmitted and propagated via PrPSc monomers and/or small aggregates. Detecting PrPSc monomers thus far has been difficult since PrPSc and PrPC have the same amino acid sequence. However, they have drastically different secondary structures with PrPC being mostly α-helical (47% α-helix, 3% β-structure) while PrPSc has significant β-structure content (17-30% α-helix, 43-45% β-structure).[2-4] This makes ion mobility mass spectrometry an ideal method to investigate this system because it separates components based on shape. There is growing evidence that the disease is both transmitted and propagated via PrPSc monomers and/or small aggregates. Detecting PrPSc monomers thus far has been difficult since PrPSc and PrPC have the same amino acid sequence. However, they have drastically different secondary structures with PrPC being mostly α-helical (47% α-helix, 3% β-structure) while PrPSc has significant β-structure content (17-30% α-helix, 43-45% β-structure).[2-4] This makes ion mobility mass spectrometry an ideal method to investigate this system because it separates components based on shape.
Funding for this research is provided by the Department for Environment, Food and Rural Affairs in the UK and is being conducted in collaboration with Jim Scrivens and Teresa Pinheiro at the University of Warwick, Department of Biological Sciences. We have recently received our first protein samples from Warwick and hope to have results soon.
References
- "Solution Structure of a 142-Residue Recombinant Prion Protein Corresponding to the Infectious Fragment of the Scrapie Isoform" Thomas L. James, He Liu, Nikolai B. Ulyanov, Shauna Farr-Jones, Hong Zhang, David G. Donne, Kiyotoshi Kaneko, Darlene Groth, Ingrid Mehlhorn, Stanley B. Prusiner, and Fred E. Cohen Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10086-10091.
- "Conversion of α-Helices into β-Sheets Features in the Formation of the Scrapie Prion Proteins" Keh-Ming Pan, Michael Baldwin, Jack Nguyen, Maria Gasset, Ana Serban, Darlene Groth, Ingrid Mehlhorn, Ziwei Huang, Robert J. Fletterick, Fred E. Cohen, and Stanley B. Prusiner Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10962-10966.
- "Secondary Structure Analysis of the Scrapie-Associated Protein PrP 27-30 in Water by Infrared Spectroscopy" Byron W. Caughey, Aichun Dong, Kolari S. Bhat, Darwin Ernst, Stanley F. Hayes, and Winslow S. Caughey Biochem. 1991, 30, 7672-7680.
- "Multiple Folding Pathways for Heterologously Expressed Human Prion Protein" Graham S. Jackson, Andrew F. Hill, Catherine Joseph, Laszlo Hosszu, Aisling Power, Jonathan P. Waltho, Anthony R. Clarke, and John Collinge Biochim. Biophys. Acta 1999, 1431, 1-13.
|

