
|

|
||
| Introduction: Xanneal is a GUI (graphic user interface) for setting up and running simulated annealing using Amber. Rather than users having to edit input files and c-shell scripts for each run, and setting up and submitting a queue script, settings can be modified from within xanneal, and then input files, the job script and queue script are all generated from within xanneal. The job can then be submitted to the queue (or optionally run in the background) from the interface. This not only cuts down on the steps that need to be followed (and remembered) each time, but also avoids the frequent errors and confusion caused by having multiple versions of scripts and input files for each user. This brief users' guide covers features of xanneal up through version 0.7 (released in November, 2005). Differences from version 0.6 include:
Type xanneal to start the interface. There must be an X server running and connected to the remote server before starting xanneal. You should see a window that looks something like this: 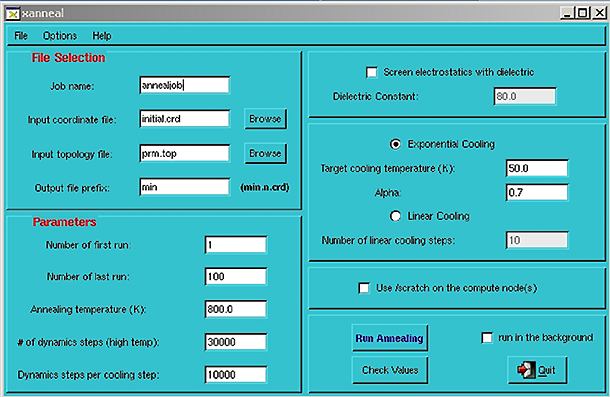 Edit options: Despite the appearance of many options, most of the defaults should be fine for a typical annealing run. The most likely options that may need to be changed are: Input coordinate file This used to always be initial.crd but now may be anything. Also, as of version 0.7 it is possible to change directory from within the interface from the file selection dialog accessed via the Browse button. In this fashion, it is possible to launch a series of jobs in different directories within the same xanneal session. This also means that it is no longer necessary to start xanneal in the directory where the job will run. Number of first and last run Change the number of last run to however many structures are desired. Change the number of first run if you are extending/restarting a previous run. Temperature Higher temperature leads to more randomized structures. Exponential or Linear cooling protocol Exponential cooling tends to find lower-energy structures. Use/scratch on the compute node(s) With this option chosen, all output files will be written to /scratch on the hard drive where the calculation is being performed, and then copied back all at once when the calculation is finished. This shouldn't normally be necessary, but we have seen instances where the performance seemed to be hindered by NFS (the network file system) and this was an obvious way to bypass the problem. Running in parallel 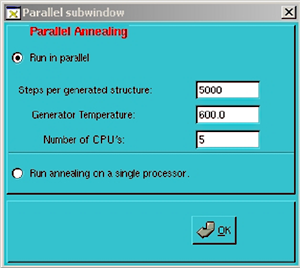 Jobs can
be broken up to run as a batch of jobs in parallel. A short amount of dynamics
is performed to generate randomized "seed" structures for each
sub-job, and then each sub-job is submitted to the queue which generates
a subset of the requested number of annealed structures. The parallel option
is turned on via the Options > Parallel
pulldown menu (right). Note that the number of CPU's does not have to be
an exact divisor of the total number of annealing steps, but it should not
be greater than the total number of steps. Jobs can
be broken up to run as a batch of jobs in parallel. A short amount of dynamics
is performed to generate randomized "seed" structures for each
sub-job, and then each sub-job is submitted to the queue which generates
a subset of the requested number of annealed structures. The parallel option
is turned on via the Options > Parallel
pulldown menu (right). Note that the number of CPU's does not have to be
an exact divisor of the total number of annealing steps, but it should not
be greater than the total number of steps.Electrostatic screening Electrostatic screening uses a dielectric constant in order to reduce the electrostatic interactions during high temperature dynamics. This allows a molecule to open up and more freely sample conformational space, especially in cases where the molecule is effectively stuck together due to large charge-charge interactions. The dielectric constant is then reduced back to the vacuum value of 1.0 during cooling, gradually turning the full electrostatic interactions back on before minimization. Restraints Restraints are usually used in cases where a coordinated ion such as Na+ is present which will often dissociate during the high temperature dynamics. Care must be taken to specify the correct atom numbers for the restraint (iat(1), iat(2)) as well as the distances r3 and r4. Note that the restraints must now be specified in a separate file whose name is specified in the input file. See the manual for details on using restraints. You can turn on restraints and then just choose File > Save all files from the pulldown menu to see how the restraints are now specified. Also, as of version 0.7, multiple restraints can be entered via the interface. Segmented hot dynamics and restart structure These two options are found in the miscellaneous subwindow accessed via the Options > Miscellaneous pulldown menu. Breaking up the hot dynamics into segments will periodically save the coordinates and velocities and restart the dynamics a number of times each annealing step. The only effect of breaking up the hot dynamics this way is that sander will recalculate the virtual box each time. The idea is that this might prevent sander from crashing when an ion moves outside the virtual box before a restraint has had a chance to pull it back in towards the molecule. Besides the small overhead associated with restarting sander for each segment, the simulation should remain unperturbed. The option to continue dynamics from the last high temperature point is also meant to decrease the likelihood of exceeding the box limits. The default is to use the minimized structure to start each annealing cycle, but this tends to give things a "kick" which can lead to dissociated ions, etc. So the most conservative way to run annealing for systems that tend to dissociate is to use segmented hot dynamics AND continue from the last high temperature point (neither of which are defaults, and both accessed via the Options > Miscellaneous pulldown menu). Saving interface settings All of the current settings in the interface can be saved for future sessions using the File > Save settings pulldown menu option. All of the default settings can be restored using the File > Restore defaults menu option. The settings are stored in the user's /home directory as ~/.xanneal.settings. Debug option This option (found at the bottom of the miscellaneous options subwindow) simply turns off all of the file cleanup in the job file(s). This will result in a large number of intermediate files being left behind, but is very useful for looking at exactly what happens during a simulation, especially when a problem is suspected in xanneal itself. Other options Most of the other options should be considered "expert" options, and the user should use care in changing them. Options specific to minimization can be accessed via the Options > Minimization pulldown menu. Any other options that are not specified in the interface can be modified by choosing File > Save all files in the pulldown menu, quitting the interface, and then editing the input files by hand. The user will then be required to submit or run the job by hand. As of version 0.7, xanneal has been extensively reworked to allow the use of xanneal as an imported python module. This will enable the use of all of the file generators in xanneal from within python scripts without the need for a GUI. Running the job: Finally, the user can have the job launched on the queue automatically by clicking on the Run annealing button. A popup window should indicate success, and you can now quit xanneal. In cases when there are no nodes available in the cluster, jobs can be run in the background on the head node by checking the run in background checkbox before clicking the Run annealing button. Please note that parallel jobs will not run this way (and don't really make sense on a single node anyway) and that running in the background will tend to slow down the head node and cause sluggish response for other users (though running a single job in the background is not a problem). Running in the background is more of a last resort for small but urgent jobs. Final words: Xanneal should be quick and intuitive to use. This should make the setting up and running of annealing jobs relatively painless, allowing users to spend their time making the parameter/topology files needed as input (the hardest task when working with Amber) and analyzing results (the fun part). Version 0.7 now has the option of saving all of the current settings in the interface. This is useful when doing a series of calculations all with the same settings. It is also possible to restore all of the program defaults. These options can be accessed via the File pulldown menu. Also, as a last resort, the settings file can be manually deleted which restores the default settings (saved as ~user/.xanneal.settings). Please pass along any suggestions for additions or changes to the interface to John Bushnell and he will try and incorporate them. Also, if you think you have encountered any errors caused by the interface (for instance in the job scripts, etc.) please report them as well. If you see errors such as "linmin failure" or "extended beyond the virtual box", it is probably a good idea to look closely at your structure and consult the Amber web site for hints as to what might be going wrong (i.e. it's probably not my fault). Enjoy! Python rant: Xanneal is written in python, an interpreted language that is easy to learn and use, and is similar to perl (but generally easier to read and understand). The graphic interface portions of xanneal utilize the PyGTK python interface to GTK+, a cross platform graphic interface toolkit written in C. The curious user is encouraged to open xanneal in a text editor (like vi) to see what the code looks like. (Note that python uses indentation instead of punctuation in most cases, unlike most other languages.) Xmin, xdyn and xanneal are the very first graphic interfaces written by me (John B.) in python, which should give you an idea of how powerful Python is. Python is ubiquitous on Linux systems, and is available for Windows, Macs and many other platforms. I hav 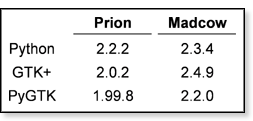 e personally
tested out xanneal on a Windows machine and it worked without modification
except for the lack of a queuing system (so I couldn't actually submit jobs). e personally
tested out xanneal on a Windows machine and it worked without modification
except for the lack of a queuing system (so I couldn't actually submit jobs).Xanneal is running smoothly on both of our Linux clusters with the versions of Python, GTK+ and PyGTK as shown in the table at left. |